Welcome to the Bioinformatics and Machine Learning Software page!
Here, you can learn about some of our recently developed bioinformatics software tools and accompanying new ML methods.
HyDRA (Hybrid Distance-score Rank Aggregation)
HyDRA is a rank aggregation tool for gene prioritization. It combines strengths of distance-based and score-based methods to provide a representative list of genes to test for its roles in a disease.
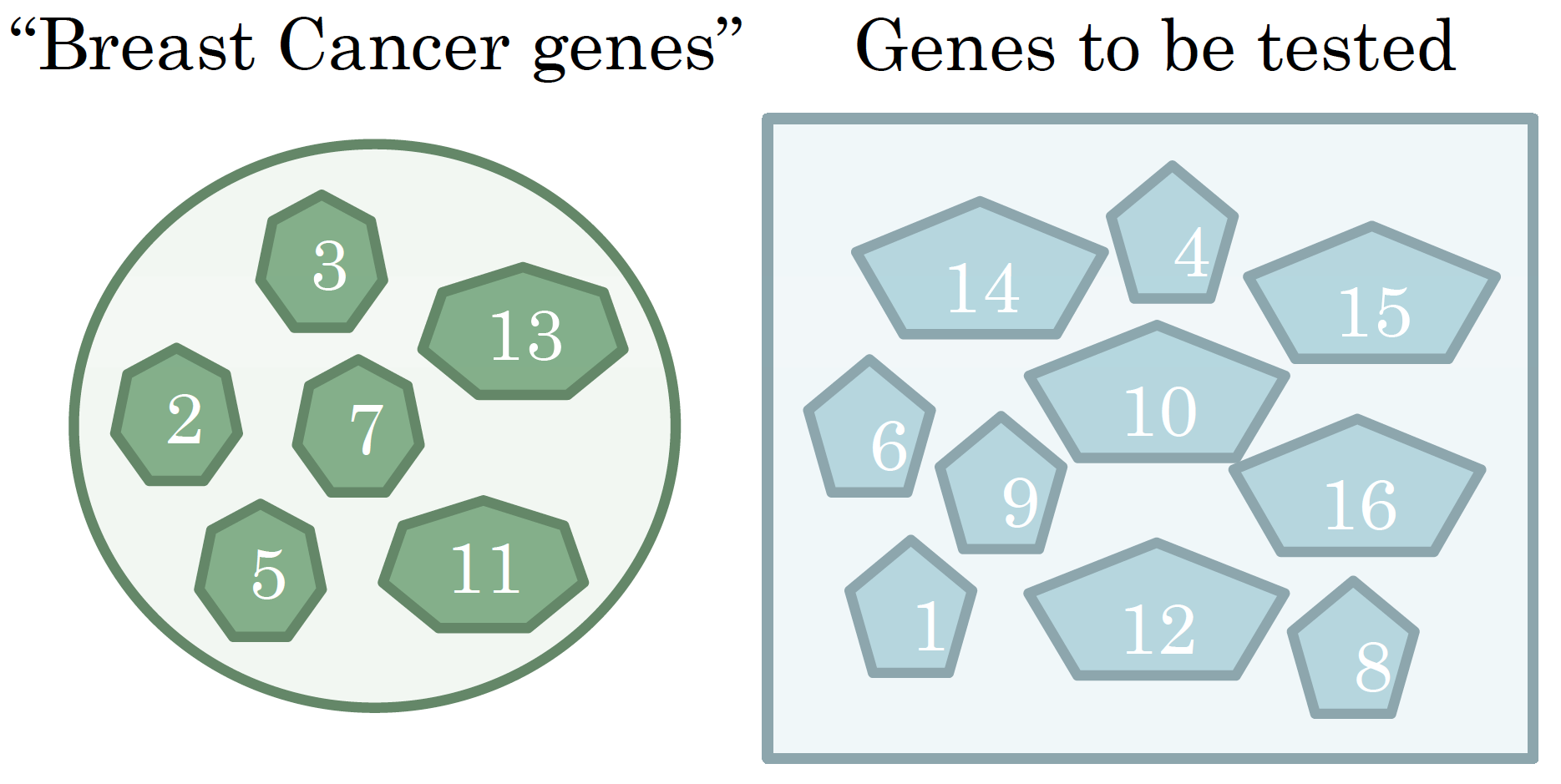
Gene Prioritization: which genes to test first? We pick the ones “most similar” to the known “Breast Cancer genes”.
C3 (Correlation Clustering Methods for Cancer Driver Gene Discovery)
C3 is a new correlation clustering method (a agnostic clustering method that can integrate various clustering constraints and multiple sources of evidence to perform provably good — i.e., constant factor approximation — clustering) specifically designed for cancer driver gene inference and pathway community analysis. It uses mutual exclusivity, coverage and gene regulatory network information to constraint an optimization problem, and TCGA data to integrate various sources of evidence (CNV, expression, methylation etc) as edge weights in the clustering problem.
An exemplary gene network.
CaSPIAN (Causal Subspace Pursuit for Inverse Engineering and Analysis of Gene Regulatory Networks)
CaSPIAN (Causal Subspace Pursuit for Inference and Analysis of Networks) is a novel algorithm for inference of causal gene interactions, based on coupling compressive sensing and Granger causality techniques. The core of the approach is to discover sparse linear dependencies between shifted time series of gene expressions using a sequential list-version of the subspace pursuit reconstruction algorithm and to estimate the direction of gene interactions via Granger-type elimination. The method is conceptually simple and computationally efficient, and it allows for dealing with noisy measurements. Its performance as a stand-alone platform without biological side-information is tested on simulated networks, on the synthetic IRMA network in Saccharomyces cerevisiae, and on data pertaining to the human HeLa cell network and the SOS network in E. coli. The results produced by CaSPIAN are compared to the results of several related algorithms, demonstrating significant improvements in inference accuracy of documented interactions. These findings highlight the importance of Granger causality techniques for reducing the number of false-positives, as well as the influence of noise and sampling period on the accuracy of the estimates. In addition, the performance of the method was tested in conjunction with biological side information of the form of sparse “scaffold networks”, to which new edges were added using available RNA-seq or microarray data. These biological priors aid in increasing the sensitivity and precision of the algorithm in the small sample regime.
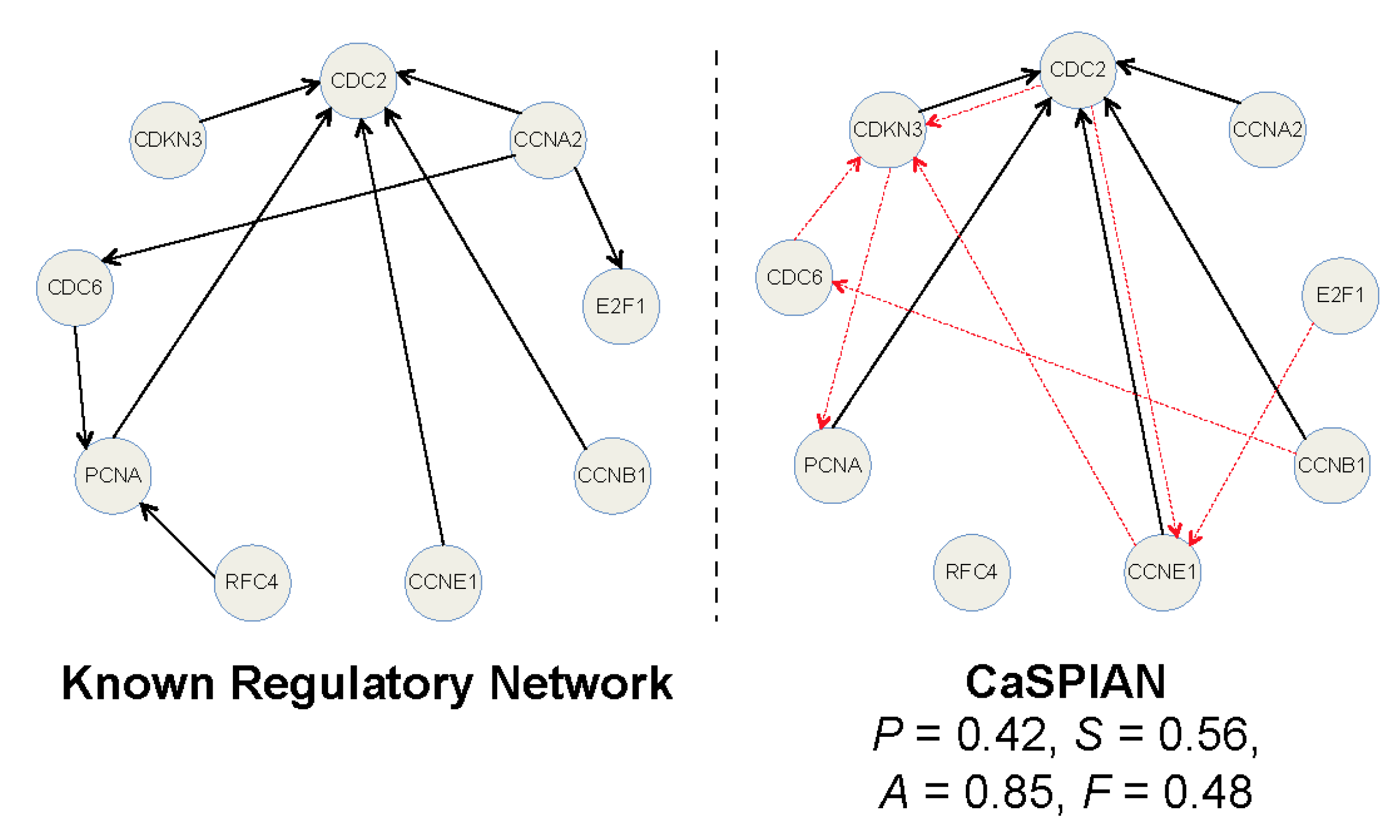
Performance of CaSPIAN.