Welcome to the NIH BD2K Targeted Software Development – Data Compression Website!
This web page is created and maintained by the Milenkovic lab.
In 2015, UIUC (Milenkovic) and Stanford (Weissman) were awarded one of the inaugural NIH BD2K Targeted Software Development Awards. This site contains software solutions for various -omics data compression problems of relevance to NIH and other bioinformatics practitioners, including:
RNA-seq Data Compression (smallWig)
ChIP-seq Data Compression (ChIPWig)
Metagenomic Data Compression (MetaCRAM)
Raw Read and Whole Genome Compression
(Under construction)
VCF File Compression
(Under construction)
Participants in this project are (in alphabetical order):
Mikel Hernandez
Minji Kim
Olgica Milenkovic
Idoia Ochoa
Vida Ravanmehr
Tsachy Weissman
Zhiying Wang
Parallel RNA-seq Data Compression with Random Access (smallWig)
smallwig is a specialized software tool for compressing RNA-seq data in the form of wig files. It offers exceptional improvements in compression rates over standard bigwig formats (for almost all tested files from the ENCODE repository, 20-fold improvements over bigwig are observed), while maintaining all the useful bigwig file properties such as summary statistics and random access.
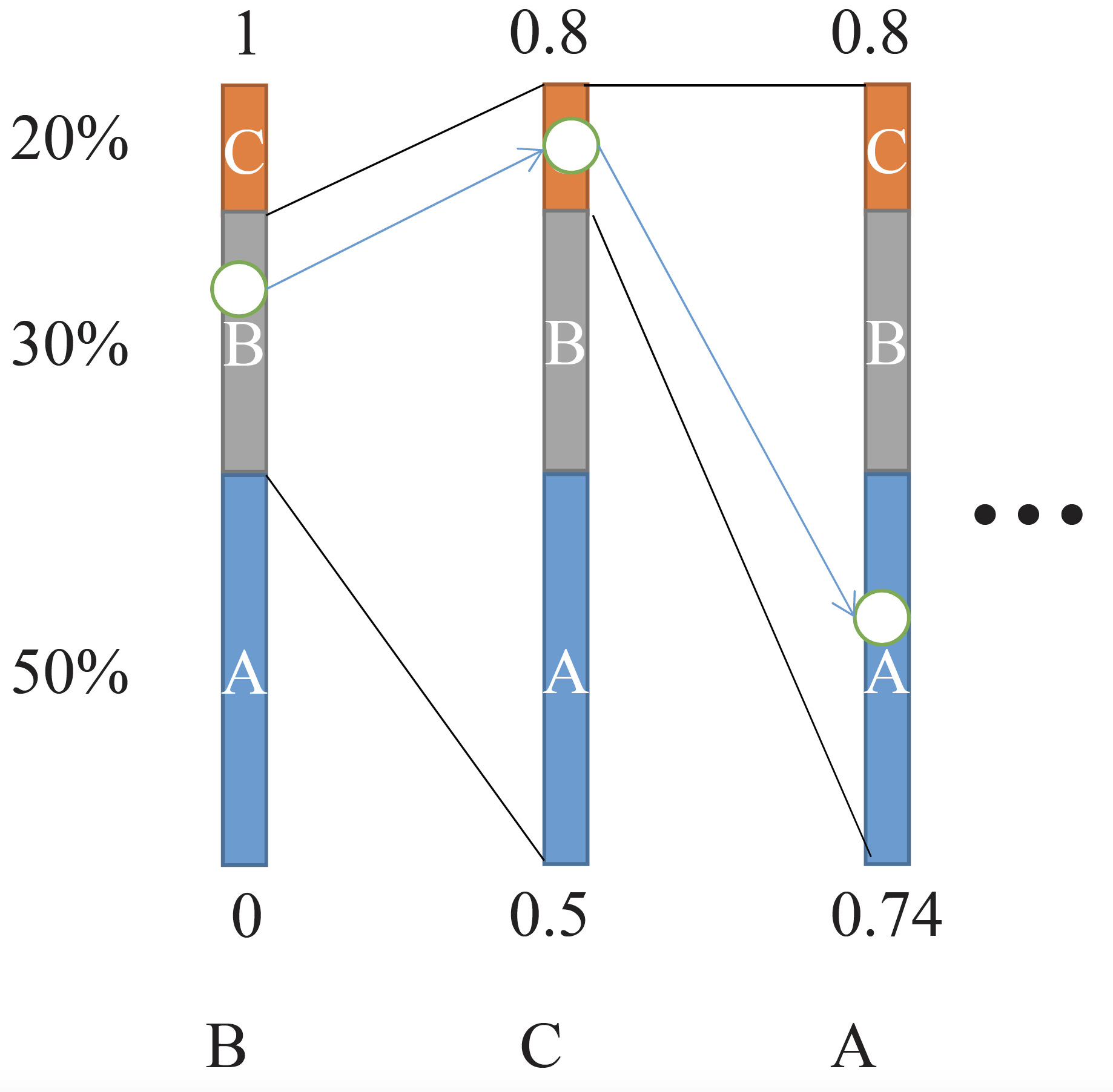
An example of arithmetic encoding method implemented in smallWig.
More information about smallwig
ChIP-seq Data Compression with Random Access (ChIPWig)
ChIPWig is a software tool which is specifically designed to compress ChIP-seq Wig files. ChIPWig combines several compression techniques including transform encoding, delta encoding, run-length encoding and arithmetic encoding. ChIPWig also enables random query functionalities and random-access in the compressed file.
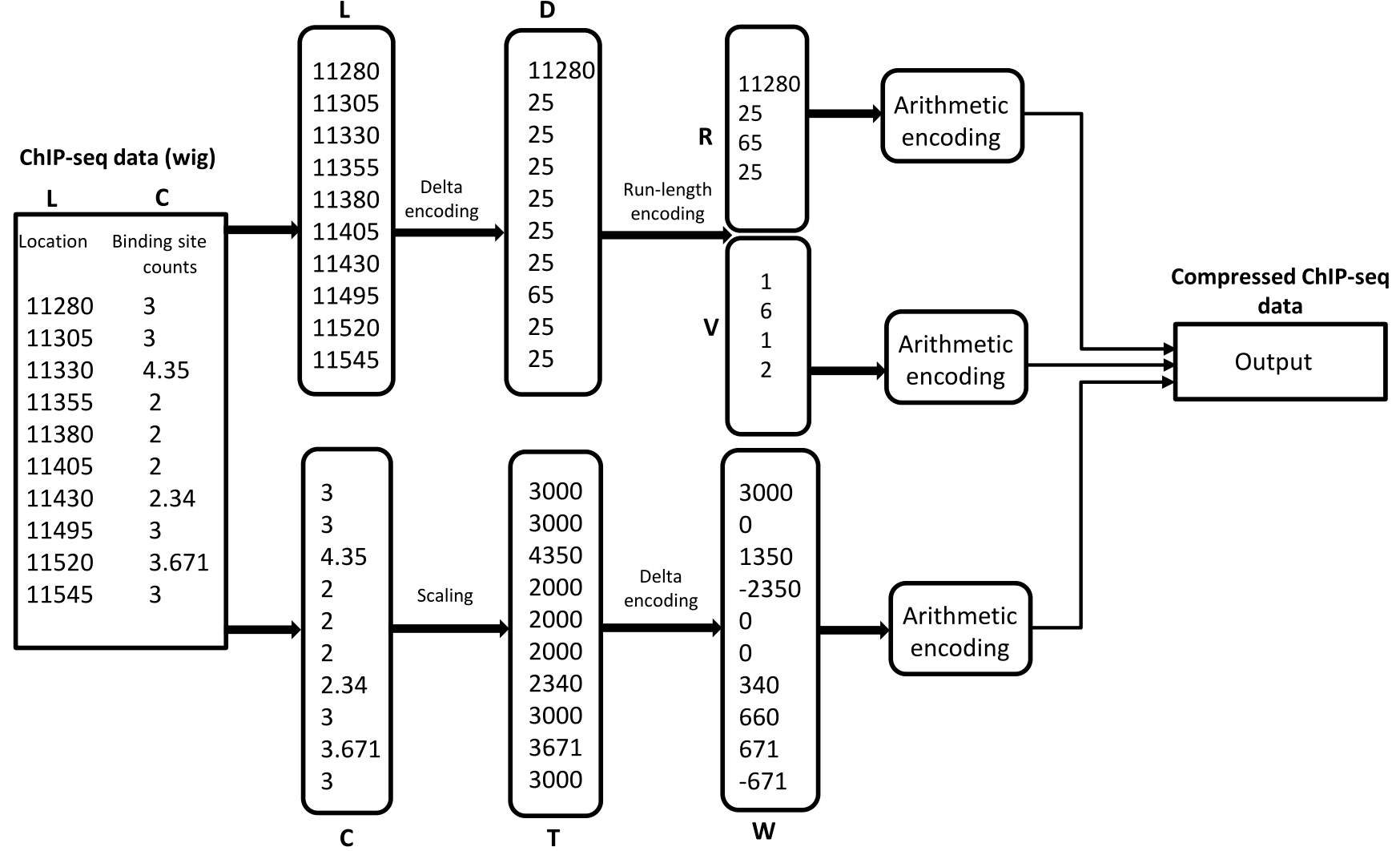
ChIPWig Compression Algorithm on a ChIP-seq Wig File
More information about ChIPWig
Metagenomic Data Compression (MetaCRAM)
MetaCRAM is a pipeline for taxonomy identification and lossless compression of FASTA-format metagenomic reads. It integrates algorithms for taxonomy identification, read alignment, assembly, and finally, a reference-based compression method in a parallel manner. As a result, the compressed file sizes were 2-13 percent of the original raw metagenomic file sizes.

The key idea is to provide a list of references for metagenomic reads, which inherently lack references. Subsequently, we encode only the small difference between the reference and our reads to achieve high compression rate. Figure credit: CRAM [EMBL]
Raw Read and Whole Genome Compression
VCF File Compression
More information about this project