Research
Graph models for Social Networks
Social networks and their analysis is an inherently interdisciplinary academic field which emerged from social psychology, sociology, statistics, and graph theory. The primary approaches to analyze social networks were mathematically formalized in the 1950s and theories and methods of social networks became pervasive in the social and behavioral sciences by the 1980s. Social network analysis is now one of the major paradigms in contemporary sociology, and is also employed in a number of other social and formal sciences. Mathematical models that have been introduced to model social networks are usually described based on concepts in Graph Theory in which individuals, groups, organizations, etc. are shown with nodes and their relationships are denoted by edges between the nodes.
One of the social network models that has been provided is based on the so called “threshold graphs” in which nodes and edges in the graph represent individuals and their connections in a network, in a way that for each node a weigh representing the wealth of each individual is assigned and there is an edge between two individuals if sum of their wealth is greater than some fixed threshold. One drawback of these graph families is that they have limited constrained generative attachment rules. To mitigate this problem, we introduced a new class of graphs termed Doubly Threshold (DT) graphs which may be succinctly described through vertex weights that govern the existence of edges via two inequalities. One inequality imposes the constraint that the sum of weights of adjacent vertices has to exceed a specified threshold. The second inequality ensures that adjacent vertices have a bounded difference of their weights. We provided a conceptually simple characterization and decomposition of DT graphs and analyzed their forbidden induced subgraphs which we compared to those of prototypical social networks. We also studied the intersection number, diameter and clustering coefficient of DT graphs.
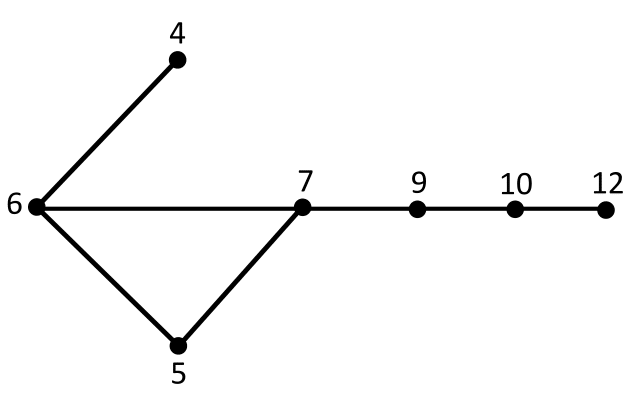
A doubly threshold graph with the thresholds 10 and 2. That is 2 vertices are adjacent if and only the sum of their weights is greater than or equal to 10, but their difference is less than 3.
ChIPWig: A State-of-the-art Compression Method for ChIP-seq Data
Chromatin immunoprecipitation-sequencing (ChIP-seq) is a technique that enables analyzing the interactions between protein and DNA using next generation sequencing technologies. ChIP-seq is an inexpensive DNA sequencing technique which combines ChIP with enormously parallel DNA sequencing to identify the binding sites of DNA-associated proteins. Specifying protein-DNA interactions and their role in regulating gene expression is of crucial importance in many biomedical applications. However, the vast volume of ChIP-seq data has caused challenges in terms of data storage, data transfer and exchange and hence increased the overall cost of data maintenance. To tackle this problem, different data compression techniques have been proposed to reduce the size of ChIP-seq files.
We propose a new low-rate compression method especially designed for ChIP-seq data. Our approach is based on source coding techniques which include transform coding, differential coding and arithmetic coding for integers and correlated real numbers. We tested our compression method on different ChIP-seq data generated by the ENCODE project. The results reveal that our method offers on average a 5-6 fold decrease in file size compared to bigWig, almost 2-fold decrease in file size compared to cWig and almost 14-fold decrease in file size compared to the original Wig files. The average size of the tested files was 1027. 75 MB in the original Wig format, 403.3125 MB in bigWig and 73.0437 MB under our compression method. Compression and decompression times of the proposed algorithm are comparable to those of bigWig.


Check-Hybrid Generalized Low-Density Parity-Check Codes
Low-density parity-check (LDPC) codes have an excellent performance under iterative message passing algorithms. However, the iterative decoders fail on some small subgraphs in the parity-check matrix of LDPC codes called trapping sets. Different methods to construct better LDPC codes or decoders have been proposed to avoid failing of decoders on small subgraphs of parity-check matrix of LDPC codes. One way to construct stronger codes is to replace all single checks of the parity-check matrix of an LDPC code with stronger and shorter error correcting codes called super checks or sub-codes. The codes constructed in this way are called generalized low-density parity-check (GLDPC) codes. If some single checks are replaced by super checks, the code is called the check-hybrid GLDPC codes.
In this project, we proposed a new approach to construct a class of check-hybrid generalized low-density parity-check (CH-GLDPC) codes which are free of small trapping sets. The approach is based on converting some selected check nodes involving a trapping set into super checks corresponding to a 2-error correcting component code. Specifically, we followed two main purposes to construct the check-hybrid codes; first, based on the knowledge of the trapping sets of the global LDPC code, single parity checks are replaced by super checks to disable the trapping sets. The second purpose is to minimize the rate loss caused by replacing the super checks through finding the minimum number of such critical checks. We also provided an analysis of error correction capability of the CH-GLDPC codes.
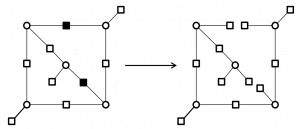
Breaking the cycles responsible for the failure of decoder by replacing super checks corresponding to a 2-error correcting component codes
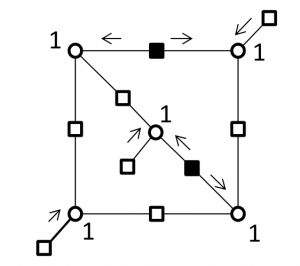
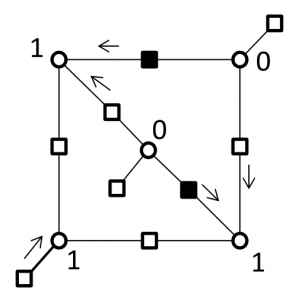
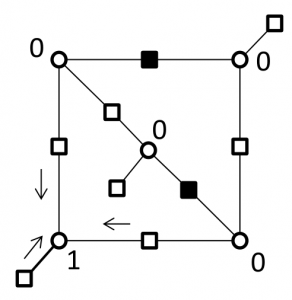
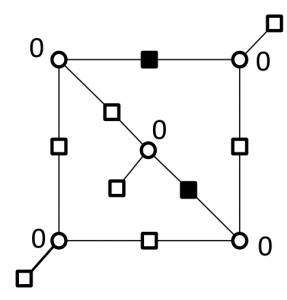
Correcting errors on a (5,3) trapping sets by replacing super checks
Gene Regulatory Network
A gene regulatory network (GRN) is a collection of molecular regulators that interact with each other and with other substances in the cell to govern the gene expression levels of proteins. These play a central role in morphogenesis, the creation of body structures, which in turn is central to evolutionary developmental biology. A gene regulatory network representing cell cycle is of special importance and has been widely studied in systems biology.
In his project, we introduced a class of finite systems models of gene regulatory networks exhibiting behavior of the cell cycle. The model is an extension of a Boolean network model. The system spontaneously cycles through a finite set of internal states, tracking the increase of an external factor such as cell mass, and also exhibits checkpoints in which errors in gene expression levels due to cellular noise are automatically corrected. We presented a 7-gene network based on Projective Geometry codes, which can correct, at every given time, one gene expression error. The topology of a network is highly symmetric and requires using only simple Boolean functions that can be synthesized using genes of various organisms. The attractor structure of the Boolean network contains a single cycle attractor.