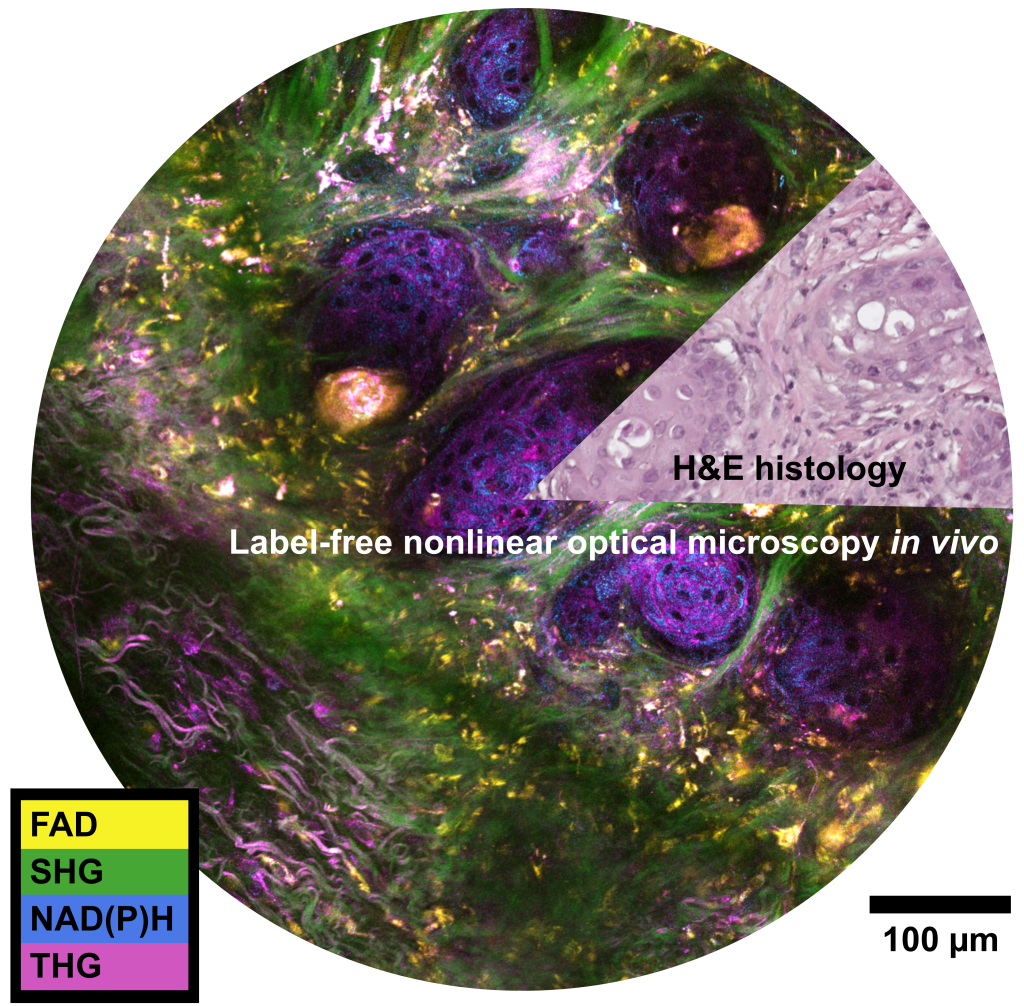
Background
My current research at the Biophotonics Imaging Laboratory is focused on development and application of novel multimodal nonlinear optical microscopes. Primarily, my focus is on label-free imaging by using endogenous (inherent) properties of biological samples to create contrast such as: multiphoton autofluorescence of naturally occurring metabolic cofactors NADH, NADPH, and FAD, coherent Raman scattering, and harmonic generation. To enable these nonlinear optical contrast mechanisms simultaneously with high spatial resolution and good penetration depth into biological samples, high peak power pulsed lasers are used for excitation. “Nonlinear” optics refers to a special set of light-matter interactions where the response of a material has a nonlinear dependence on the input intensity, such as two-photon excited fluorescence.
Here are some papers that are useful resources for learning more about label-free nonlinear optical microscopy:
- Coherent Raman scattering microscopy in biology and medicine (C Zhang et al., 2015)
- Fluorescence lifetime imaging microscopy: fundamentals and advances in instrumentation, analysis, and applications (R. Datta et al., 2020)
- Intravital imaging by simultaneous label-free autofluorescence-multiharmonic microscopy (S You et al., 2018)
- Label-free optical metabolic imaging in cells and tissues (I Georgakoudi, K Quinn, 2023)
- Nonlinear magic: multiphoton microscopy in the biosciences (WR Zipfel, RM Williams, WW Web, 2004)
- The phasor approach to fluorescence lifetime imaging analysis (M Digman et al, 2008)
- Two-photon excitation fluorescence microscopy (PTC So et al., 2000)
Technology development
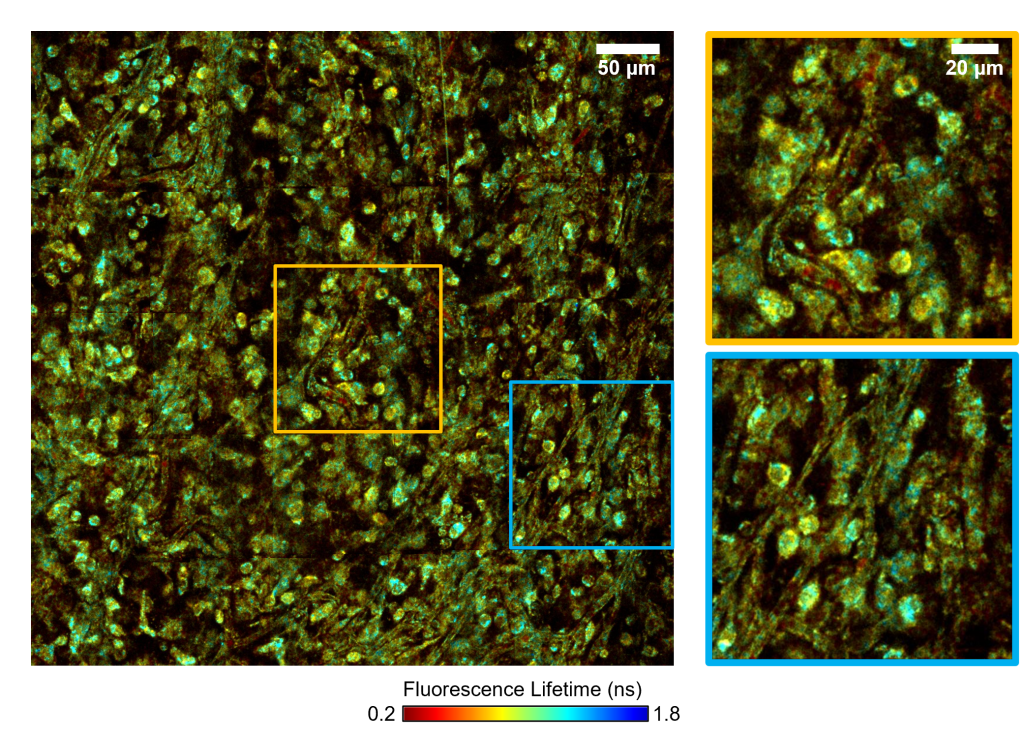
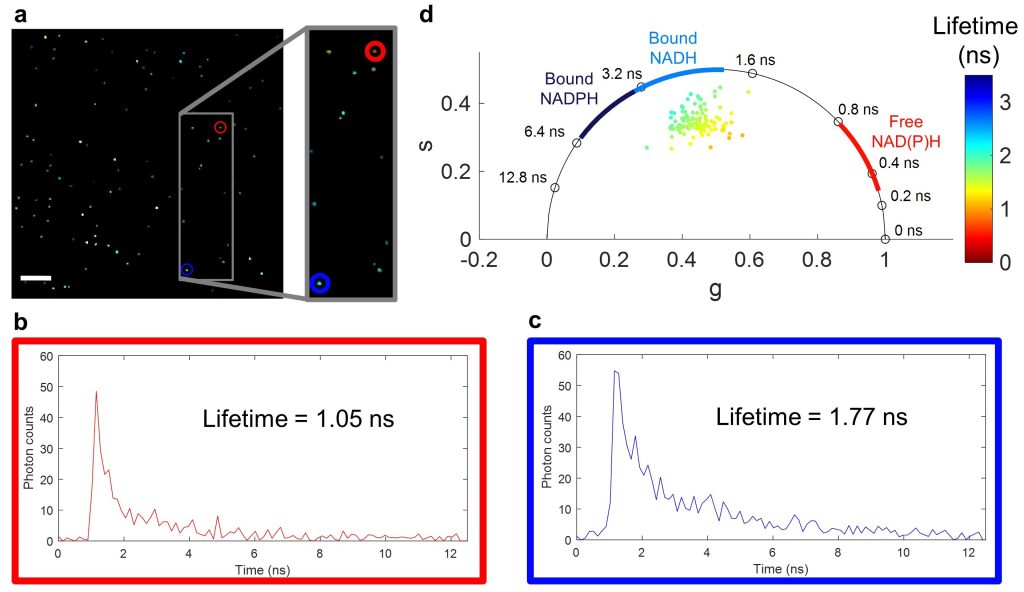
Fluorescence lifetime imaging microscopy (FLIM) examines the ns-scale differences in fluorescence emission from fluorophores, which can differ based on fluorophore environment (pH, ion concentration, protein binding). For label-free imaging, autofluorescent cofactors NAD(P)H and FAD can be imagined and quantified based on their fluorescence intensity and lifetime in order to make inferences about the metabolic state of cells and tissues.
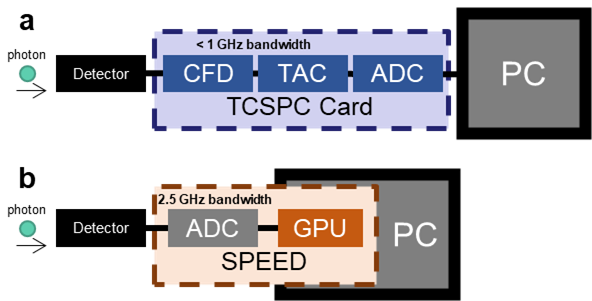
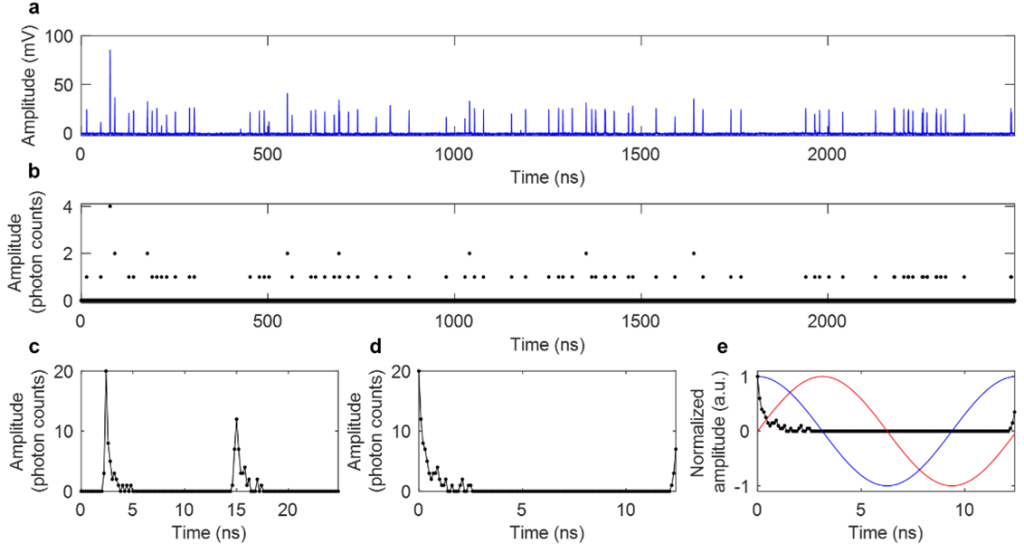
One barrier to widespread use of FLIM is that it generally suffers from slow acquisition due to the “dead time” (time after one photon is collected that another cannot be counted) that stems from the limited bandwidth of photon-counting and time-tagging analog electronics. To overcome this, I came up with Single- and multi-photon PEak Event Detection (SPEED), a method to count photons on directly digitized data, thus bypassing the slower electronics (Sorrells et al., 2021).
I also investigated differences in the commonly use photomultiplier tubes (PMTs) vs. hybrid photodetectors (HPDs), and found that with HPDs, the SPEED method enables accurate photon counting for multiphoton FLIM at photon rates up to 223% (Sorrells et al., 2022). This work enables faster and higher-throughput FLIM which can be used to investigate new biological processes at a fast timescale with previously impossible precision.
Cancer cell death
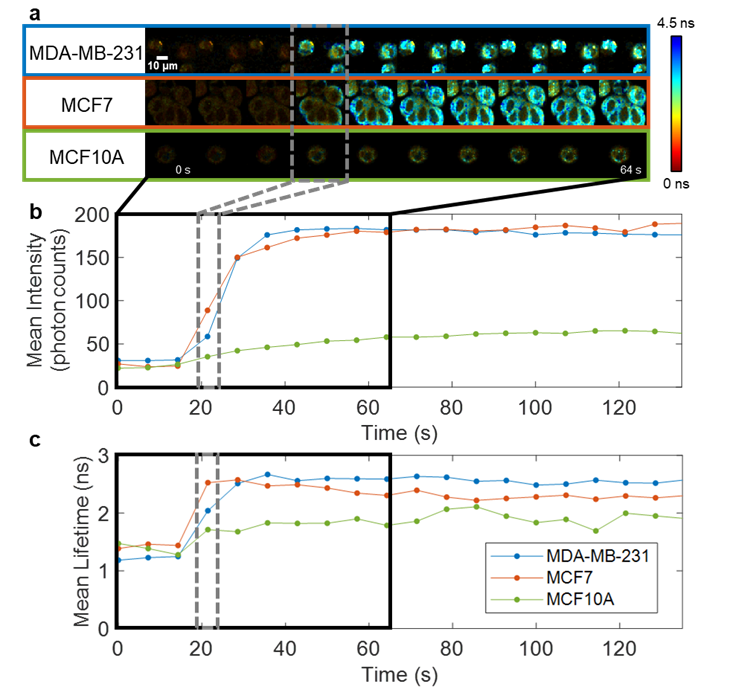
Fast FLIM acquisition is essential for studying rapid cellular metabolic dynamics. Apoptosis of cancer cells is a key goal of chemotherapy, and when cancer cells evade apoptosis they can contribute to cancer recurrence and/or metastasis. My hope is to study the cellular metabolic dynamics of cancer cells undergoing in the presence of apoptotic agents to better understand the mechanisms of apoptosis and chemoresistance. The previously described FLIM technology has been applied for looking at programmed cell death, termed apoptosis, and found that cellular NAD(P)H fluorescence intensity and lifetime show intense increases upon apoptosis induction (Sorrells et al., 2023; Bower et al., 2019). I also worked on a collaborative study supported by the Mayo Clinic & Illinois Alliance for Technology-Based Healthcare using optical metabolic imaging to characterize the effect of chemotherapy on human patient-derived xenografts (PDX) from pancreatic ductal adenocarcinoma (PDAC) implanted into mice, which showed that tumors resistant to chemotherapy initially showed a response similar to those of responsive tumors, but deviated after five weeks (Park and Sorrells et al., 2023).
Extracellular vesicles
Extracellular vesicles (EVs) are small membrane-bound particles that are released by cells. Recently, there has been a huge increase in interest in EVs due to their potential in therapeutics and diagnostics. Furthermore, EVs have been implicated in a variety of diseases and disorders, such as cancer metastasis. However, EVs are hard to study due to their small size, heterogeneity, and lack of university markers. To overcome this, researchers are BIL discovered that label-free nonlinear optical microscopy could be used to visualize and characterize EVs and that they have distinct and meaningful optical signatures (You et al., 2019). Due to my interest in FLIM, I developed methods to image and characterize EVs with multiphoton FLIM of NAD(P)H to provide information about NAD(P)H-protein interactions within EVs (Sorrells et al., 2021). Since these microscopy methods are label-free and non-invasive, they can also be performed on live cells and tissues, allowing for direct comparison between EVs and their sources and for visualization of EVs in situ. One interesting finding from my work so far is that EVs most closely resemble the NAD(P)H autofluorescence signature of the cytosol of their parents cells but show a much more broad/heterogenous profile (Sorrells et al., 2021).