smallwig: Parallel and Random-Access RNA-seq Data Compression Software
RNA-seq Data
Rna-Sea (sometimes referred to whole transcriptome shotgun sequencing), is a next-generation sequencing method for determining the expression level (i.e., level of RNA, small RNA, miRNA, etc.) in a given sample. As such, it is an indispensable tool for many comparative studies and for elucidating cellular phenomena such as alternative gene splicing, post-transcriptional modifications, and gene fusion.

Image credit: Sonika Tyagi, Australian Genome Research Facility.
What is smallwig?
smallwig is a specialized compression algorithm for wig files that contain RNA-seq data. It may be used in the archival mode, in which case the file is compressed using computationally intense methods such as context-tree weighting, leading to near-optimal compression rates; and in the standards mode, which enables access to summary statistics of blocks of data, random access and fast parallel compression/decompression features.
smallwig offers significantly better compression rates than standard bigwig, gzip and wig methods, in most cases more than an order of magnitude (most test examples were files from the ENCODE project). The compression rate and running time both in standard mode and random query mode are shown in the following figures.
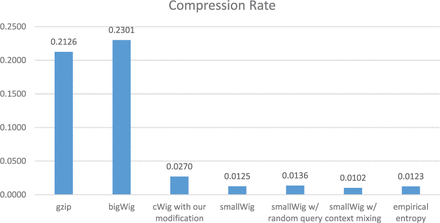
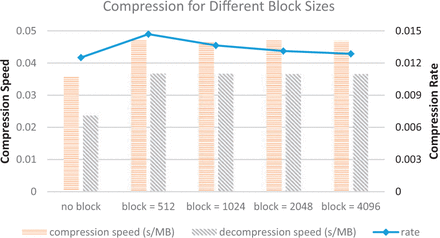
How to cite this paper:
Zhiying Wang, Tsachy Weissman, and Olgica Milenkovic, “smallWig: parallel compression of RNA-seq WIG files”, Bioinformatics, 32(2), pp.173-180, 2016.
Contact
For questions, please contact Zhiying Wang (zhiying@uci.edu) and Olgica Milenkovic (milenkov@illinois.edu).
How to access smallwig?
You may access the source code for smallwig and the README file from github, here.
Usage
The README file instructions are summarized below.
==============================================
To compress a file, please use:
wig2smallwig [InputFile] [OutputFile]
options:
-m [N=0..9, uses 3 + 3*2^N MB memory, decompress should use same N]
context mixing
-r [B, encode block size from 8 to 32]
random access and encode by blocks of size 2^B
-p [total number of processes]
parallel realization, only available if -r is enabled and -m is disabled
To decompress a file, please use:
To use:
smallwig2wig [InputFile] [OutputFile]
options:
-s [ChrmName (e.g. chr1)] [Query Start (integer)] [Query End (integer)]
subsequence query
-p [total number of processes]
parallel realization, only available if -s is disabled and -r is enabled in encoding
==============================================
==============================================
1. Compress a file using standard setup:
$ wig2smallwig in.wig out.swig
2. Compress a file allowing random query in the future:
$ wig2smallwig in.wig out.wig -r 9 13
3. Compress a file allowing random query in the future, and use 4 parallel processors:
$ wig2smallwig in.wig out.wig -r 9 13 -p 4
4. Compress a file as much as possible for archive purposes using the maximum amount of memory:
$ wig2smallwig in.wig out.wig -m 9
5. Decompress a whole file:
$ smallwig2wig in.swig out.wig
6. Decompress a file only in chr1, start from location 300, end at location 500:
$ smallwig2wig in.swig out.wig -s chr1 300 500
7. Decompress a file with 4 parallel processors:
$ smallwig2wig in.swig out.wig -p 4