Lossless Compression Tool for Metagenomic Reads
What is MetaCRAM?
MetaCRAM is a pipeline for taxonomy identification and lossless compression of FASTA-format metagenomic reads. It integrates algorithms for taxonomy identification, read alignment, assembly, and finally, a reference-based compression method in a parallel manner.
Why use MetaCRAM?
We live in a “Big Data era” in which the amount of information is overwhelming for current storage and processing capacity. Metagenomic data is no exception: the volume of sequencing data is increasing rapidly, requiring a compression tool for long-term archival storage. MetaCRAM precisely addresses this problem and its performance was evaluated on various metagenomic samples from the NCBI Sequence Read Archive, suggesting 2- to 4-fold compression ratio improvements compared to gzip. On average, the compressed file sizes were 2-13 percent of the original raw metagenomic file sizes. Compression ratios of this order will provide for tremendous storage savings.
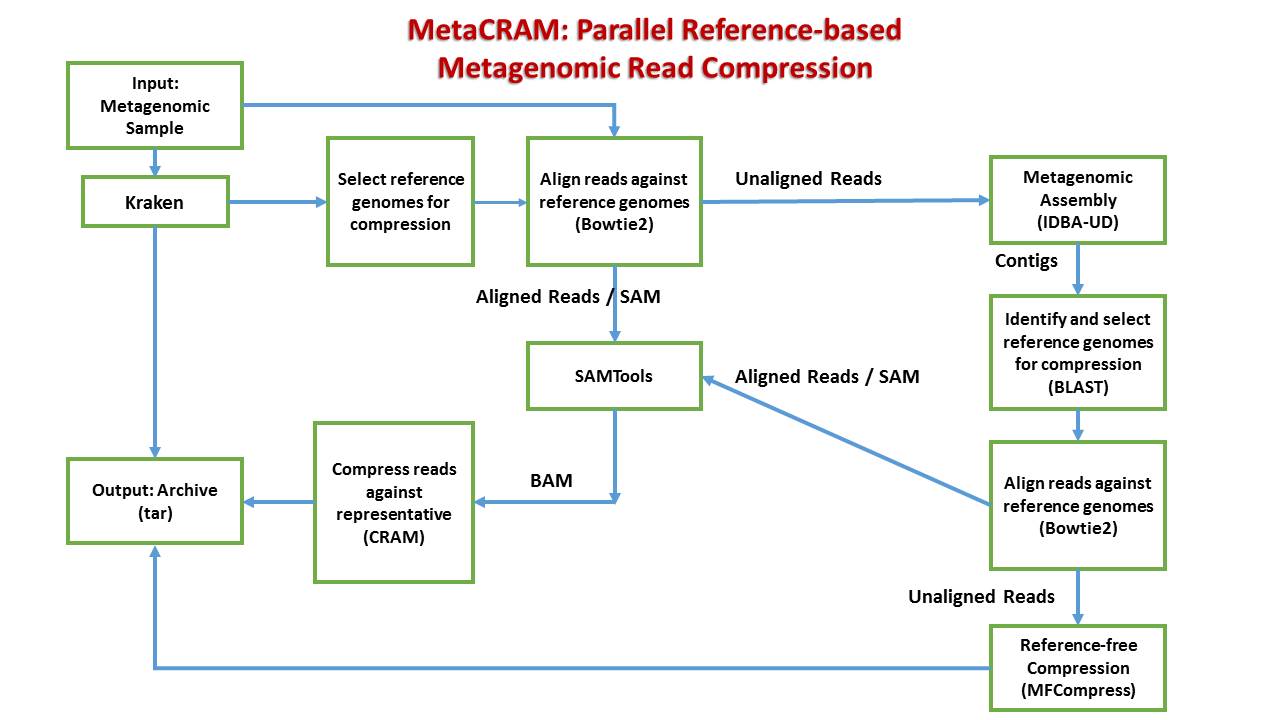
Figure. Block diagram of MetaCRAM.
How do I download MetaCRAM?
You may access MetaCRAM’s source code, installation guideline and README in the Github repository.
System Requirement
We tested MetaCRAM on a linux machine with Intel Core i5-3470 CPU at 3.2 GHz, with a 16 GB RAM.
Usage
After following the installation guideline from our Github repository, use the following commands to run MetaCRAM.
Compression
perl MetaCram.pl –compress –output <output directory> –paired <path to reads> –<exGolomb, huffman, golomb>
Example:
[shared3]$ perl MetaCram.pl –compress –output /shared3/MetaCRAM_SRR359032_Huffman –paired /shared3/SRR359032_1.fasta /shared3/ SRR359032_2.fasta –huffman
Decompression
perl MetaDeCram.pl –input <path to folder containing the Round1 and Round2 folders>
Example:
[shared3]$ perl MetaDeCram.pl –input /shared3/MetaCRAM_processedSRR359032_Huffman/MetaCRAM
(*–paired is optional)
(*<> indicates a choice)
For more information on options, use:
$ perl MetaCram.pl –help
Reference
“MetaCRAM: An Integrated Pipeline for Metagenomic Data Processing and Compression”.
M. Kim, X. Zhang, J.G. Ligo, F. Farnoud, V.V. Veeravalli, O. Milenkovic.
BMC Bioinformatics, 17(1), (2016). [link]
Contact
Minji Kim (mkim158@illinois.edu) and Olgica Milenkovic (milenkov@illinois.edu)